Co to jest amyloidoza dziedziczna?
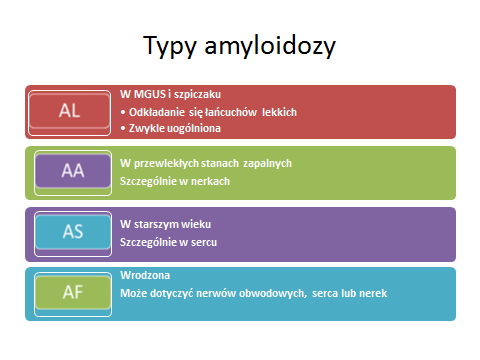
Dziedziczna amyloidoza (z angielskiego: hereditary amyloidosis), przez niektórych nadal nazywana rodzinną (familial amyloidosis, AF), rozwija się na skutek mutacji zachodzącej w materiale genetycznym danego chorego w czasie jego życia lub jest dziedziczona od jednego z rodziców. Najczęściej dotyczy genu dla transtyretyny (TTR), która jest białkiem produkowanym w wątrobie, transportującym tyroksynę (hormon tarczycy) i witaminę A we krwi. Wtedy jest określana jako amyloidoza transtyretynowa typ zmutowany (ATTRm). W wyniku mutacji białko to uzyskuje nieprawidłową budowę i odkłada się w organach jako amyloid. Główną lokalizacją złogów białkowych jest serce, poza tym niszczone są nerwy obwodowe. Inne, rzadziej spotykane formy dziedzicznej amyloidozy dotyczą białek takich jak: łańcuch alfa fibrynogenu (AfibA), gelsolina (Agel), białka lizosomów (Alys). Pierwsze objawy pojawiają się dopiero w średnim wieku.
Dziedziczna amyloidoza transtyretynowa – ATTRm
Dziedziczna amyloidoza transtyretynowa (ATTRm) to rzadka, postępująca, nieodwracalna i śmiertelna forma amyloidozy, powodowana przez mutacje w genie kodującym białko transtyretyny (TTR).
Transtyretyna z mutacją V30M – główna przyczyna ATTRm
Transtyretyna jest białkiem transportowym dla hormonu tarczycy (tyroksyny) i witaminy A. W warunkach fizjologicznych krąży w surowicy lub osoczu jako białko rozpuszczalne. 95% transtyretyny powstaje w wątrobie, <5% w mózgu i oku.
Prawidłowy kompleks transtyretyny składa się z czterech połączonych ze sobą podjednostek. Mutacje w genie TTR prowadzą do powstania wariantów, tworzących niestabilne tetrametry. Dotychczas zidentyfikowano ponad 100 różnych mutacji w genie kodującym TTR. Najczęstsza z nich jest mutacja V30M. Po oddysocjowaniu, niepoprawnie sfałdowane podjednostki polimeryzują i gromadzą się w postaci włókien amyloidowych w nerwach obwodowych i autonomicznych.
Obraz kliniczny ATTRm
Pierwsze objawy ATTRm z mutacją V30M występują zwykle w 3-4 dekadzie życia. U pacjentów z mutacjami innymi niż V30M objawy występują po 50. roku życia. Należą do nich:
- Zajęcie nerwów obwodowych: zaburzenia czuciowo-ruchowe w obrębie kończyn dolnych, objawiające się upośledzeniem odczuwania temperatury oraz bólu, hiperalgezją, a także osłabieniem i zanikiem mięśni w miarę postępu upośledzenia czuciowego. Zwyrodnienie aksonalne postępuje w kierunku dystalno-proksymalnym.
- Zajęcie układu autonomicznego: jeden lub kilka z następujących objawów: występujące naprzemiennie zaparcia i biegunki, hipotonia ortostatyczna, zaburzenia erekcji, obrzęk i niezamierzona utraty masy ciała. Mogą być widoczne w momencie wystąpienia pierwszych oznak choroby lub pojawić się po upływie 1–2 lat od wystąpienia początkowych objawów czuciowo-ruchowych.
- Zajęcie układu sercowo-naczyniowego, manifestujące się zaburzeniami rytmu serca.
Oczekiwana długość życia nieleczonych pacjentów wynosi średnio 10 lat od pojawienia się objawów. W tym czasie dochodzi do postępującego pogorszenia funkcji czuciowych, ruchowych oraz ze strony układu autonomicznego.
Występowanie choroby
ATTRm jest niezwykle rzadka. Występuje prawie na całym świecie, a najbardziej powszechna jest w populacji portugalskiej, japońskiej i szwedzkiej – 1 na 1000 do 1 na 10 000 osób. W pozostałych populacjach występuje z częstością poniżej 1 przypadku na 10 000 osób.
Rozpoznanie ATTR-FAP – polineuropatii w ATTRm
Rozpoznanie ATTR-FAP (ang. familial amyloid polyneuropathy) może być wyzwaniem, gdyż jej objawy są zbliżone do objawów innych polineuropatii. Kluczowym etapem podczas diagnozowania jest więc uwzględnienie ATTR-FAP na pierwszym miejscu w diagnostyce różnicowej.
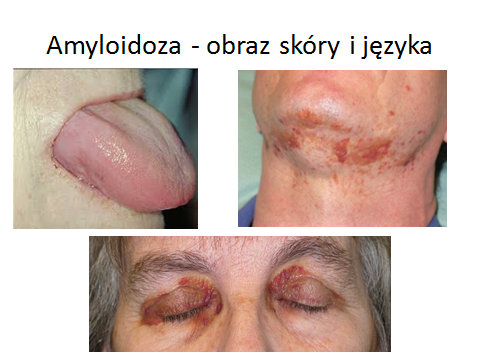
Rozpoznanie stawiane jest na podstawie potwierdzenia występowania złogów amyloidowych TTR oraz identyfikacji mutacji w genie kodującym TTR. Amyloid identyfikuje się na podstawie biopsji np. z podskórnej tkanki tłuszczowej brzucha, narządu zajętego ATTR-FAP (np. nerwy, serce, nerki) lub innego, niezajętego miejsca, np. dziąseł, śluzówki odbytnicy, ślinianek. Dokładne określenie mutacji w badaniach genetycznych jest istotne dla rokowania i leczenia. U pacjentów z pozytywnym wywiadem rodzinnym pod kątem ATTR-FAP badanie genetyczne może być wystarczające do postawienia diagnozy.
Obciążenie dla pacjentów
Utrzymującemu się upośledzeniu funkcji neurologicznych związanemu z ATTR-FAP towarzyszy istotne obniżenie jakości życia i wpływa szczególnie na niezależność i samoocenę pacjenta. Pacjenci, jak również ich rodziny, często wymagają porady, wsparcia i informacji, aby być w stanie poradzić sobie z wpływem, jaki choroba ma na ich życie.
Leczenie ATTRm
ATTR można leczyć farmakologicznie, metodami inwazyjnymi lub objawowo.
Jedynym preparatem zarejestrowanym w leczeniu ATTR-FAP jest Tafamidis (Vyndaquel®). Terapia tym lekiem polega na stabilizacji tetrameru transtyretyny. Lek ten wykazuje poprawę funkcji neurologicznych i stanu odżywienia w porównaniu do placebo. Lek został zarejestrowany w Europie w 2011 do terapii na wczesnym etapie choroby – u pacjentów z objawami polineuropatii, ale chodzących w pełni samodzielnie.
Leczenie farmakologiczne innymi preparatami jest możliwe np. przez udział w badaniach klinicznych.
Obecnie trwają zaawansowane badania kliniczne z lekami oligonukleotydowymi (patisiran, inotersen), których wstępne wyniki są bardzo obiecujące.
Metodę inwazyjną – przeszczep wątroby, stosuje się zwykle, gdy terapia tafamidisem okazuje się nieskuteczna. W razie wystąpienia kardiomiopatii może być wykonany także przeszczep serca.
Rejestr THAOS – ogólnoświatowe źródło informacji dotyczących ATTR-FAP
THAOS (Transthyretin Amyloidosis Outcomes Survey) to trwający 10 lat rejestr obserwacyjny, prowadzony metodą strategii porównań podłużnych, do którego dane mogą wprowadzać wszyscy lekarze prowadzący leczenie pacjentów z amyloidozą transtyretynową.
Rejestr THAOS ma na celu:
- Lepsze zrozumienie naturalnego przebiegu amyloidozy transtyretynowej, w tym zmienności i progresji wrodzonych oraz nabytych postaci choroby.
- Lepsze zrozumienie zależności pomiędzy genotypem i fenotypem w przypadku amyloidozy transtyretynowej.
- Porównanie progresji choroby i całkowitego czasu przeżycia w przypadku pacjentów z amyloidozą transtyretynową poddanych i niepoddanych przeszczepowi wątroby.
- Ocena metod leczenia, które mogą być korzystne w przypadku pacjentów z amyloidozą transtyretynową.
- Stworzenie międzynarodowej społeczności specjalistów medycznych opracowujących zalecenia dotyczące klinicznego postępowania w przypadku pacjentów z amyloidozą transtyretynową.











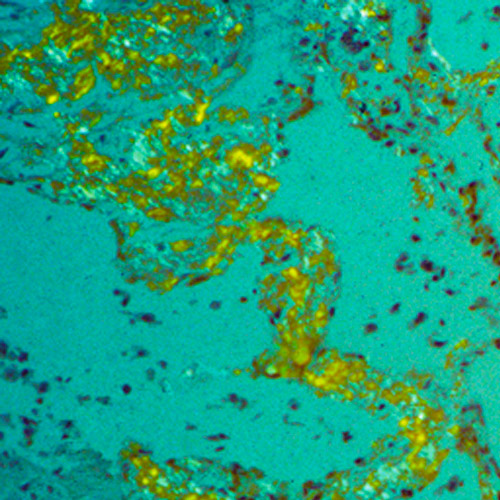
 Biopsja tkanki tłuszczowej skóry brzucha, źródło: www.amyloid.nl
Biopsja tkanki tłuszczowej skóry brzucha, źródło: www.amyloid.nl
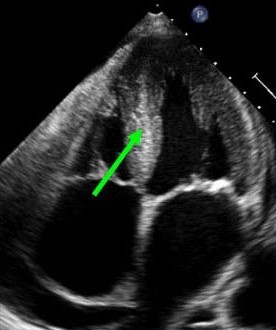 Obraz amyloidozy serca w badaniu echokardiograficznym
Obraz amyloidozy serca w badaniu echokardiograficznym