Główny cel diagnostyki to wykrycie depozytów amyloidu w tkankach na podstawie badania bioptycznego (pobranie tkanki za pomocą specjalnej igły). Najłatwiej materiał do badania uzyskać z biopsji tkanki tłuszczowej skóry brzucha. Jest to zabieg bezpieczny, wykonywany w znieczuleniu miejscowym, w warunkach ambulatoryjnych (nie jest konieczny pobyt w oddziale szpitalnym). Można też wykonać biopsję błony śluzowej jamy ustnej czy odbytnicy, a także narządów podejrzanych o nacieczenie amyloidem, na przykład nerek. Jednak niektóre organy, jak serce, bardzo rzadko są badane w ten sposób z uwagi na wysokie ryzyko powikłań przy zabiegu.

Wizyta u lekarza ma na celu ustalenie prawidłowej diagnozy. Z tego względu niezbędne jest wykonanie szeregu badań i testów.

Amyloidozy można i powinno się leczyć, jednak postępowanie jest zależne od podtypu choroby. W przypadku amyloidozy AA należy zwalczać stan zapalny, który spowodował jej rozwój. W amyloidozie AL leczeniem z wyboru jest chemioterapia, na temat której więcej informacji zamieszczono poniżej. Nie wolno zapominać o leczeniu wspomagającym – bardzo ważnym we wszystkich typach schorzenia, nakierowanym na ochronę funkcji uszkodzonych narządów.

Europejskie Stowarzyszenie pacjentów chorych na szpiczaka (Myeloma Patients Europe, MPE) opracowało szereg informacji dla pacjentów i organizacji stowarzyszających grupy pacjentów dotyczących dostępnych metod leczenia szpiczaka plazmocytowego jak również innych ważnych problemów związanych z chorobą.

O ile amyloidoza transtyretynowa nabyta dotyczy głównie serca, to postać genetycznie uwarunkowana uszkadza wiele narządów, głównie nerwy obwodowe i ser-ce, ale również oczy i nerki. Choroba ta rozwija się u osób dorosłych, z wiekiem zachorowania od ok. 4-tej dekady życia do późnej starości. Do jej rozwinięcia wystarczy jeden wadliwy gen, ale nie zawsze musi się on ujawnić, mówimy w tym przypadku o tzw. niepełnej penetracji genu. Przyczyny różnego wieku zachorowania i objawów nawet w tej samej rodzinie nie są znane.

Badanie izotopowe SPECT stało się kluczową techniką w identyfikacji pacjentów z amyloidozą ATTR. W badaniu tym wykorzystuje się radioizotop technetu 99m w połączeniu ze znacznikami klasycznie stosowanymi w badaniu układu kostnego.

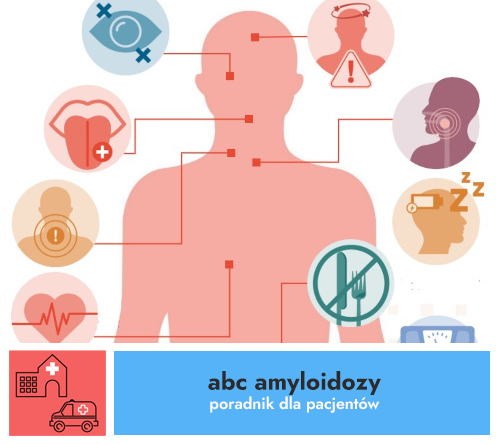
Termin amyloidoza odnosi się do grupy chorób polegających na gromadzeniu się w organizmie nieprawidłowo uformowanego białka, zwanego amyloidem. Amyloidoza jest poważną chorobą, która może doprowadzić do zagrażających życiu uszkodzeń narządów. Amyloidoza występuje bardzo rzadko. Istnieje wiele rodzajów amyloidoz. Nazwę choroby tworzy się dodając do członu „amyloidoza” nazwę białka, które odkłada się w narządach, np. amyloidoza łańcuchów lekkich (w skrócie AL). Stanowi ona ok 75% wszystkich amyloidoz, a jej szacowana częstość to 9 przypadków na milion mieszkańców/rok. Szacuje się jednak, że z powodu charakterystyki objawów amyloidozy, które są przypisywane innym, bardziej powszechnym jednostkom chorobowym, jej częstość pozostaje niedoszacowana.

Amyloidoza łańcuchów lekkich (zwana również amyloidozą AL) jest bardzo rzadką chorobą. Pojawia się, kiedy nieprawidłowe komórki plazmatyczne w szpiku kostnym produkują źle uformowane białka – łańcuchy lekkie immunoglobulin, które w takiej postaci noszą nazwę amyloidu. Włókna amyloidu dostają się do krwiobiegu i odkładają w postaci złogów w wielu tkankach i ważnych narządach, powodując objawy chorobowe.
Amyloidoza łańcuchów lekkich (zwana również amyloidozą AL) jest bardzo rzadką chorobą. Pojawia się ona, kiedy nieprawidłowe komórki plazmatyczne w szpiku kostnym produkują źle uformowane łańcuchy lekkie immunoglobulin, które w takiej postaci noszą nazwę amyloidu.

Amyloidoza AA jest spowodowana odkładaniem się w przestrzeni zewnątrzkomórkowej amyloidu, którego prekursorem jest białko surowiczego amyloidu AA. Zaliczane jest ono do białek ostrej fazy, których stężenie w surowicy wzrasta wraz z rozwojem i utrzymywaniem się stanu zapalnego. Typowymi chorobami, których powikłaniem może być amyloidoza AA są przewlekłe stany zapalne o nieznanej przyczynie, choroby autoimmunologiczne, nowotwory oraz przewlekłe infekcje.

Zachęcamy do zapoznania się z poradnikiem kierowanym do pacjentów chorych na amyloidozę i ich rodzin.

Rozmowa ze Zbigniewem Pawłowskim, prezesem Stowarzyszenia Rodzin Pacjentów z Amyloidozą.

Rozmowa z prof. Przemysławem Mitkowskim, kierownikiem Pracowni Elektroterapii Serca i Kliniki Kardiologii Uniwersytetu Medycznego im. Karola Marcinkowskiego w Poznaniu.

26 października 2023 r. obchodzimy Trzeci Światowy Dzień Amyloidozy. Jest to rzadka grupa chorób, których elementem wspólnym jest nieprawidłowe gromadzenie się białka (amyloidu) w różnych narządach i tkankach, prowadząc do ich uszkodzenia i zaburzeń funkcjonowania.

Amyloidoza to grupa chorób, których wspólnym mianownikiem jest pozakomórkowe odkładanie się nierozpuszczalnego białka, amyloidu, w różnych narządach. W nabytej amyloidozie transtyretynowej – inaczej amyloidozie ATTR – jednym z podtypów choroby dotyczącym w większości mężczyzn po 60. roku życia, głównym miejscem gromadzenia się amyloidu jest serce. Pacjenci cierpiący na to schorzenie przez lata mogą nie być jej świadomi, a diagnozę otrzymać dopiero, kiedy pojawi się u nich niewydolność serca.

Amyloidoza transtyretynowa to ultrarzadka choroba, która, jak wskazują specjaliści, w kardiologii jest dość unikatowa. Znana jest ona już od 150 lat, a do niedawna rozpoznawana była dopiero po zgonie pacjenta. Dziś mamy możliwości diagnostyki tej choroby oraz zatrzymania jej rozwoju dzięki już rozpoznanym lekom. Na złoty standard leczenia w Polsce nadal czeka kilkaset pacjentów.
- « Poprzednia
- 1
- 2
- 3
- 4
- 5
- 6
- …
- 9
- 10
- 11
- Następna »