
Drugi typ amyloidozy ATTR to postać dziedziczna, związana z amyloidogennymi mutacjami genu transtyretyny. Występuje endemicznie m.in. w Portugalii. W Polsce jest bardzo rzadka, rozpoznano ją dotychczas zaledwie u kilkunastu pacjentów.
Transtyretyna jest białkiem produkowanym w wątrobie, którego główną rolą fizjologiczną jest transport tyroksyny i retinolu. Przyczyny powstawania amyloidu z transtyretyny nie są dokładnie poznane. Wiemy, że proces odkładania się amyloidu w sercu prawdopodobnie trwa wiele lat, zanim rozwinie się obraz kardiomiopatii i pojawią się objawy uszkodzenia serca. Z tego względu tak ważne jest szybkie rozpoznanie choroby i wczesne rozpoczęcie terapii przyczynowej, zanim dojdzie do nieodwracalnego uszkodzenia serca.
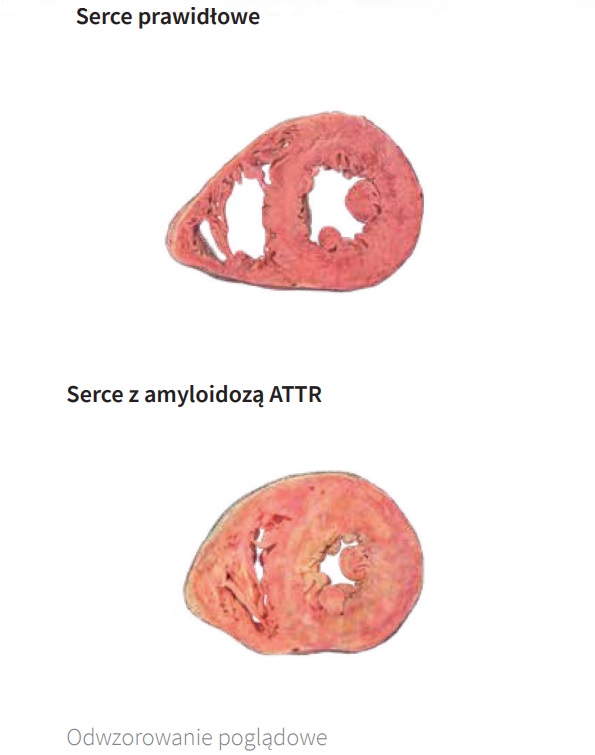
Amyloid może gromadzić się również w innych narządach, ale najczęstszą prezentacją kliniczną amyloidozy transtyretynowej jest kardiomiopatia z obrazem klinicznym niewydolności serca. Chorzy są często kierowani do Oddziału Kardiomiopatii z błędnym rozpoznaniem sarkomerowej kardiomiopatii przerostowej. Amyloidoza transtyretynowa najczęściej przebiega pod maską kardiomiopatii przerostowej o profilu restrykcyjnym, a w zaawansowanej fazie jako niewydolność serca z zachowaną frakcją wyrzutową (tzw. rozkurczowa niewydolność serca). I to właśnie w tych dwóch populacjach pacjentów należy jej przede wszystkim szukać.
Amyloidoza jest chorobą znaną od połowy XIX wieku. Wówczas były to jednak rozpoznania sekcyjne, nie potrafiliśmy też różnicować typów amyloidozy. Długo była uważana za chorobę idiopatyczną, o nieznanej przyczynie, której nie rozpoznaje się za życia i nie ma możliwości leczenia. Wraz z postępem wiedzy medycznej mamy coraz większą możliwość poznawania przyczyn chorób, co jest kluczowe dla znalezienia skutecznych metod leczenia, powstały również nowe metody diagnostyki.
Amyloidoza transtyretynowa serca jest jednym z dwóch rodzajów amyloidozy. Aby ją rozpoznać musimy najpierw wykluczyć amyloidozę z łańcuchów lekkich immunoglobulin. Jest ona wprawdzie chorobą o zupełnie innej przyczynie, wywodzącą się z komórek plazmatycznych szpiku, spokrewnioną ze szpiczakiem mnogim, ale jej prezentacja kardiologiczna i to, co widzimy w badaniach obrazowych serca, jest w zasadzie identyczne w obu typach amyloidozy. Dopiero bardziej szczegółowa diagnostyka pozwala nam postawić właściwe rozpoznanie, a jest to kluczowe, bo amyloidozę łańcuchów lekkich coraz skuteczniej leczą hematolodzy.
Wczesne rozpoznanie ATTR-CM jest niezwykle istotne, ponieważ rokowanie pogarsza się gwałtownie wraz z dalszym odkładaniem się złogów amyloidowych, późniejszą postępującą dysfunkcją narządów i znacznym obniżeniem jakości życia.
Zaawansowane stadium ATTR-CM u nieleczonych pacjentów wiąże się z poważnymi powikłaniami kardiologicznymi i gorszą medianą przeżycia. Po rozpoznaniu mediana przeżycia nieleczonych pacjentów z ATTR-CM i objawami niewydolności serca wynosi w przybliżeniu od 2 lat do 3,5 roku. Wczesne, trafne rozpoznanie ATTR-CM może przynieść korzyści w opiece nad pacjentem i umożliwić osiągnięcie lepszych wyników leczenia.
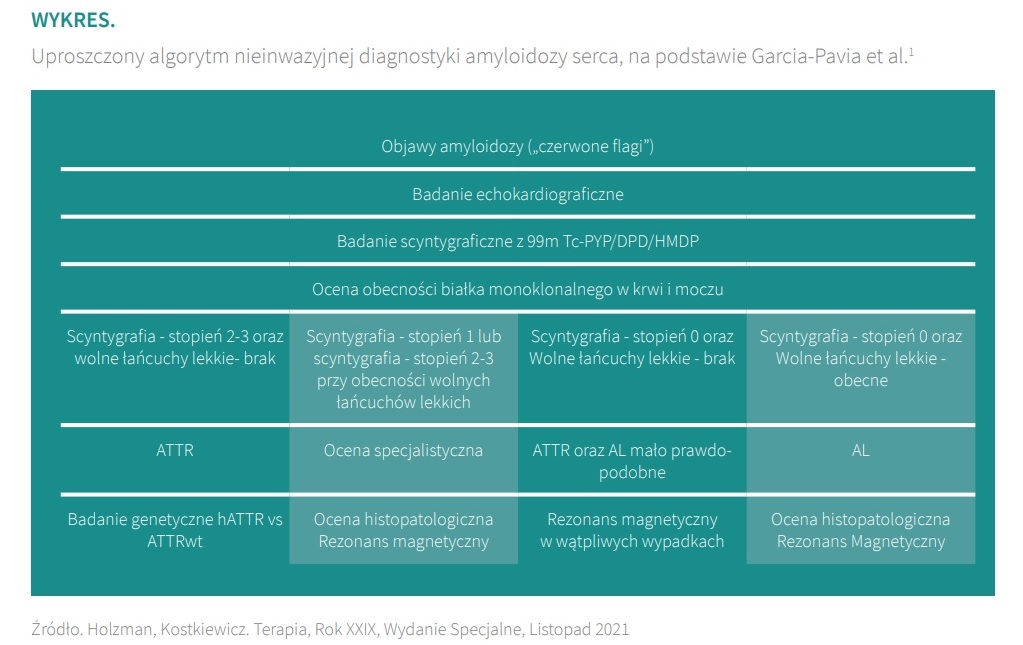
Algorytm diagnostyczny amyloidozy transtyretynowej zmienił się z pierwotnie inwazyjnego na nieinwazyjny. Historycznie amyloidozę rozpoznawało się za pomocą biopsji tkanki tłuszczowej i ta metoda jest nadal stosowana w diagnostyce amyloidozy hematologicznej, z łańcuchów lekkich immunoglobulin. Natomiast w amyloidozie transtyretynowej czułość biopsji tkanki tłuszczowej wynosi tylko kilkanaście procent. Zachęcałbym więc lekarzy do tego, żeby zamiast wykonywania biopsji zaczynali od łatwo dostępnej diagnostyki nieinwazyjnej, czyli badania echokardiograficznego i, w miarę możliwości, rezonansu magnetycznego serca. Przełomem w diagnostyce amyloidozy transtyretynowej było wprowadzenie diagnostyki izotopowej. Badaniem, które pozwala z bardzo wysoką dokładnością rozpoznać ten typ amyloidozy, jest scyntygrafia kości z zastosowaniem dwufosfonianu znakowanego technetem (99mTc-DPD). Badanie to od wielu lat wykorzystywane jest w onkologii. Dopiero niedawno potwierdzono, że jedyną jednostką chorobową, w której ten izotop w specyficzny sposób gromadzi się w sercu ‒ zamiast wkościach ‒ jest właśnie amyloidoza transtyretynowa. To badanie na razie jest w Polsce dostępne w nielicznych pracowniach medycyny nuklearnej, ponieważ znacznik ten nie jest zarejestrowany do badania serca. U każdego chorego, u którego podejrzewamy amyloidozę, musimy przeprowadzić różnicowanie z amyloidozą łańcuchów lekkich immunoglobulin. Współcześnie mamy do dyspozycji łatwo dostępny laboratoryjny test diagnostyczny, wymagający tylko pobrania próbki krwi i moczu, w którym szukamy występowania białka monoklonalnego i oceniamy poziom wolnych łańcuchów lekkich. Ujemny wynik testu w połączeniu z dodatnim wynikiem scyntygrafii jest wystarczający do postawienia rozpoznania amyloidozy transtyretynowej.
W diagnostyce amyloidozy serca głównym problemem jest wciąż mała świadomość istnienia tej choroby. Przede wszystkim należałoby więc zacząć od szerzenia wiedzy na jej temat wśród lekarzy, w tym zwłaszcza kardiologów. Kolejnym ważnym czynnikiem jest czujność diagnostyczna, a więc znajomość sygnałów alarmowych, sugerujących rozwój choroby (tzw. ang. red flags). Spełnienie tych dwóch warunków jest kluczowe.
Podejrzenie ATTR-CM i potrzebę dalszych badań sugerują następujące objawy kliniczne, a zwłaszcza ich współwystępowanie:
- niewydolność serca z zachowaną frakcją wyrzutową (HFpEF) u pacjentów zazwyczaj w wieku powyżej 60. roku życia,
- nietolerancja standardowych metod leczenia niewydolności serca, takich jak inhibitory konwertazy angiotensyny, antagoniści receptora angiotensyny i beta-adrenolityki,
- niezgodność pomiędzy woltażem zespołów QRS na elektrokardiogramie (EKG) a grubością ściany lewej komory (LV),
- echokardiografia wykazująca pogrubienie ściany lewej komory, ze współistniejącymi cechami restrykcji,
- rozpoznanie schorzeń ortopedycznych, w tym zespołu cieśni nadgarstka, zwężenia kanału kręgowego w odcinku lędźwiowym, zerwania ścięgna mięśnia dwugłowego, lub artroplastyka stawu biodrowego i kolanowego,
- zaburzenia układu nerwowego, w tym polineuropatia i zaburzenia autonomicznego układu nerwowego, m.in. dolegliwości żołądkowo-jelitowe lub niewyjaśniona utrata masy ciała.2
Obecnie do dyspozycji mamy na etapie wstępnej diagnostyki stosunkowo proste, powszechnie dostępne metody diagnostyki nieinwazyjnej, obejmujące badanie elektrokardiograficzne, echokardiograficzne i wreszcie coraz szerzej dostępne biomarkery sercowe (NT-proBNP i troponina T). Upowszechnienie wiedzy na temat amyloidozy wśród lekarzy jest warunkiem wcześniejszego rozpoznawania tej choroby. Kilka lat temu opublikowano wyniki badania przeprowadzonego w USA, z którego wynikało, że zaledwie u 10 proc. pacjentów amyloidozę rozpoznawał pierwszy lekarz specjalista. Z kolei co najmniej jedna czwarta pacjentów była badana przez co najmniej pięciu lekarzy różnych specjalności, zanim pojawiło się rozpoznanie amyloidozy. A wczesne rozpoznanie jest bardzo ważne, gdyż jest to choroba postępująca, cały czas odkłada się amyloid w mięśniu sercowym i z chwilą kiedy serce jest w dużym stopniu zajęte amyloidem, zaczyna się bardzo szybka progresja objawów niewydolności serca. W późnej fazie choroby nie ma już możliwości leczenia. Ostatnio pojawiają się nowe leki, są perspektywy leczenia przyczynowego, więc tym bardziej tak ważne jest wczesne rozpoznanie. W Oddziale Kardiomiopatii Narodowego Instytutu Kardiologii w Warszawie – Aninie w ostatnich kilku latach rozpoznaliśmy amyloidozę transtyretynową u 39 pacjentów. Jak dotąd liczba nowych rozpoznań zwiększa się powoli. Oceniamy, że pacjentów kwalifikujących się do leczenia w Polsce może być obecnie około pięćdziesięciu. Jeśli świadomość występowania tej choroby i znajomość jej cech charakterystycznych wśród lekarzy będzie się poprawiała, to liczba rozpoznań zacznie rosnąć Na razie jednak w Polsce jest to choroba nie tyle rzadka, co raczej ultrarzadka.
Pacjenci z amyloidozą często źle tolerują typowe leczenie objawowe – leki kardiologiczne stosowane w leczeniu nadciśnienia tętniczego i niewydolności serca ‒ inhibitory konwertazy angiotensyny i betablokery. To może być pomocne w samym rozpoznawaniu – u osób starszych, które są leczone tymi lekami, w pewnym momencie ciśnienie się obniża, pojawia się nietolerancja stosowanego leczenia, co jest sygnałem, że możemy mieć do czynienia z amyloidozą. Ze względu na postępującą dysfunkcję autonomicznego układu nerwowego w zaawansowanej fazie amyloidozy, leki te bardzo nasilają hipotonię i zasłabnięcia w tym mechanizmie. Czasami betablokery są potrzebne w kontroli rytmu serca, ale trzeba je stosować ostrożnie. Natomiast podstawowymi lekami objawowymi w leczeniu niewydolności serca u chorego z amyloidozą transtyretynową są diuretuki pętlowe.
Nadzieję dla chorych na amyloidozę serca stwarza leczenie przyczynowe tafamidisem. Tafamidis został zarejestrowany do leczenia kardiomiopatii w przebiegu amyloidozy transtyretynowej przez Agencję Żywności i Leków (FDA) w USA w 2019 r., a w 2020 r. przez Europejską Agencję Leków (EMA). Lek stabilizuje tetramery transtyretyny, czyli zapobiega tworzeniu włókien amyloidu, działając w sposób selektywny, nie ma działania przeciwzapalnego. Tafamidis spowalnia postęp choroby zatrzymując produkcję amyloidu, który już się nie odkłada w narządach, zwłaszcza w sercu. Hamuje postęp amyloidozy transtyretynowej, ale nie usuwa złogów amyloidu, które już są w sercu. Dlatego jest niesłychanie ważne, aby był zastosowany na jak najwcześniejszym etapie choroby. Przypomnijmy, że przed wprowadzeniem tej terapii średni czas przeżycia chorych od momentu rozpoznania choroby wynosił od 2 do 6 lat, w zależności od zaawansowania choroby w chwili rozpoznania. W Polsce tafamidis nie jest jeszcze refundowany, jest stosowany w ramach terapii charytatywnej, finansowanej przez producenta, głównie w dwóch wyspecjalizowanych ośrodkach: w Oddziale Kardiomiopatii Narodowego Instytutu Kardiologii w Warszawie – Aninie oraz w Szpitalu im. Jana Pawła II w Krakowie. Łącznie w Polsce leczonych jest ok. 40 pacjentów, w tym 26-ciu w Oddziale Kardiomiopatii. Programwczesnego dostępu wykazał, że najmłodszy zakwalifikowany pacjent miał 47 lat, a najstarszy 79 lat. U tych chorych, u których tafamidis został zastosowany w fazie umiarkowanego zaawansowania choroby, nasze wstępne doświadczenia są dobre, a postęp choroby został zatrzymany. Nasze obserwacje są wprawdzie krótkie, ale wyniki są bardzo obiecujące. W zaawansowanej fazie badań klinicznych są obecnie leki działające w innym mechanizmie, które hamują ekspresję genu odpowiedzialnego za produkcję transtyretyny.
W amyloidozie serca bardzo ważna jest kompleksowa opieka nad pacjentem i jego rodziną od początku procesu diagnostyki, przez leczenie, aż po rehabilitację. Wzorcowym przykładem ośrodka klinicznego świadczącego kompleksową opiekę dla kilku tysięcy pacjentów w Wielkiej Brytanii jest Narodowe Centrum Amyloidozy (The National Amyloidosis Centre, NAC) w Royal Free Hospital i University College London (UCL).
Narodowe Centrum Amyloidozy to w pełni zintegrowana placówka badawcza i kliniczna, która od ponad 30 lat znajduje się w czołówce badań i leczenia wszystkich aspektów amyloidozy, a od 1999 r. na zlecenie brytyjskiej Narodowej Służby Zdrowia (NHS) zapewnia ogólnokrajową, wysoce specjalistyczną opiekę kliniczną. Działalność Centrum obejmuje badania molekularne, genetyczne, biochemiczne, fizjologiczne, kliniczne nad nowymi lekami, eksperymentalne i patologiczne, a także optymalizację terapii i rehabilitacji. W wielu z tych dziedzin istnieją rozległe powiązania współpracy z naukowcami, klinicystami i przemysłem. Podstawową misją badawczą Narodowego Centrum Amyloidozy jest wyjaśnienie podstawowych mechanizmów patobiologicznych w celu poprawy diagnostyki i leczenia amyloidozy. Multidyscyplinarną opiekę kliniczną zapewniają specjaliści z zakresu reumatologii, immunologii, nefrologii, neurologii i kardiologii.3 W Polsce, zgodnie z zaleceniami Planu dla Chorób Rzadkich, powinny powstać specjalistyczne referencyjne ośrodki kliniczne świadczące wielodyscyplinarną i kompleksową opiekę nad chorymi na amyloidozę serca. W pełni przygotowane do pełnienia takiej roli są obecnie Narodowy Instytut Kardiologii w Warszawie – Aninie oraz Klinika Chorób Serca i Naczyń Szpitala im. Jana Pawła II w Krakowie.

Piśmiennictwo:
- Garcia-Pavia P., Rapezzi C., Adler Y. i wsp.: Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. European Heart Journal 2021, 42: 1554–1568.
- Kardiomiopatia w przebiegu amyloidozy transtyretynowej (ATTR-CM). Diagnostyka obrazowa ATTR-CM za pomocą scyntygrafii. PP-VYN-POL-0064, Pfizer Rare Diseases.
- Philip N Hawkins, FMedSci, Marianna Fontana, MD PhD, Julian D Gillmore, MD PhD, The UK National Amyloidosis Centre: The National Amyloidosis Centre at the Royal Free Hospital and University College London is the world’s largest amyloidosis centre with almost 1500 patient referrals annually, European Heart Journal, Volume 40, Issue 21, 1 June 2019, Pages 1661–1664, https://doi. org/10.1093/eurheartj/ehz346